|
| | ||||||
| Main | News | Description | Download | Contacts | Help | Blog | |
| Order | Update | Gallery | About Us | Links | Forum | Citation | |
|
Chemcraft Gallery - tools for molecular dynamics researchers
Chemcraft reads GROMACS .xtc and .trr files, GAMESS .irc files, and .xyz files with multiple trajectory steps. A set of tools is provided for convenient analysis and processing of the data with molecular dynamics trajectories:
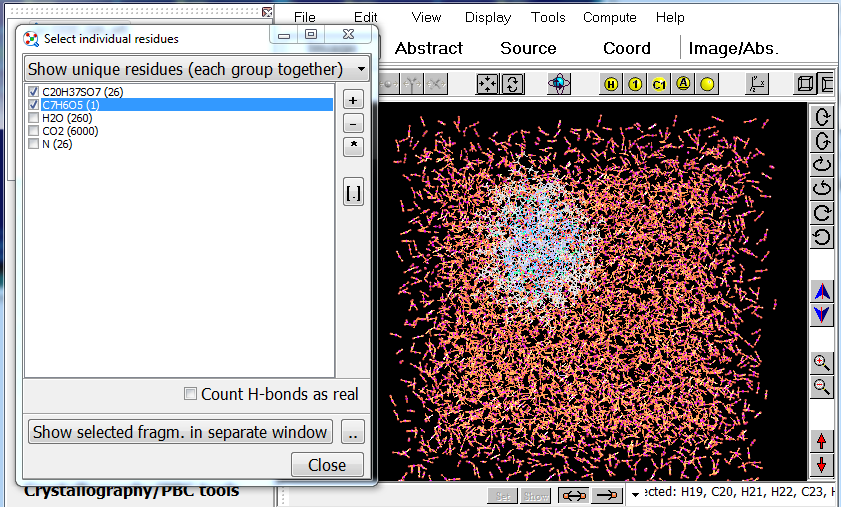
1. The tool "Tools/Divide current structure..." can split your system into individual residues (bonded groups of atoms, molecules). You can select individual residues or groups of residues with same molecular formula; also, you can check whether H-bonds are considered as "real" in this utility, so you can select individual dimers, trimers, etc:
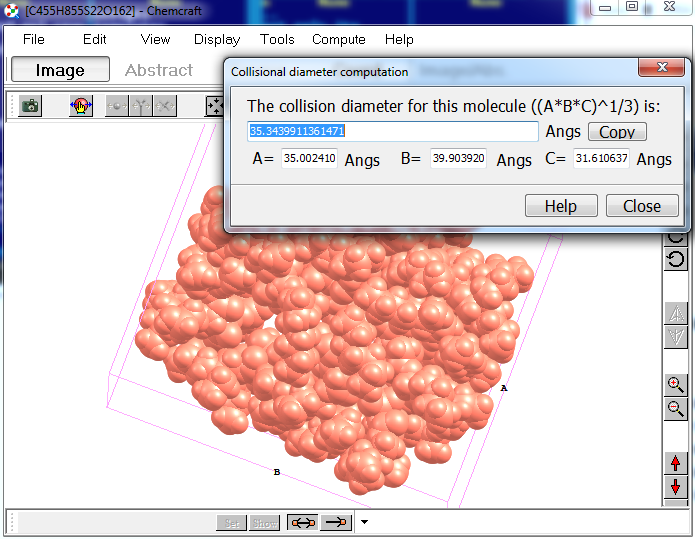
2. Also, when working with residues, you can compute the size of a residue as the A, B, C parameters of a parallelepiped with the lowest volume fitting all atoms in the molecule:
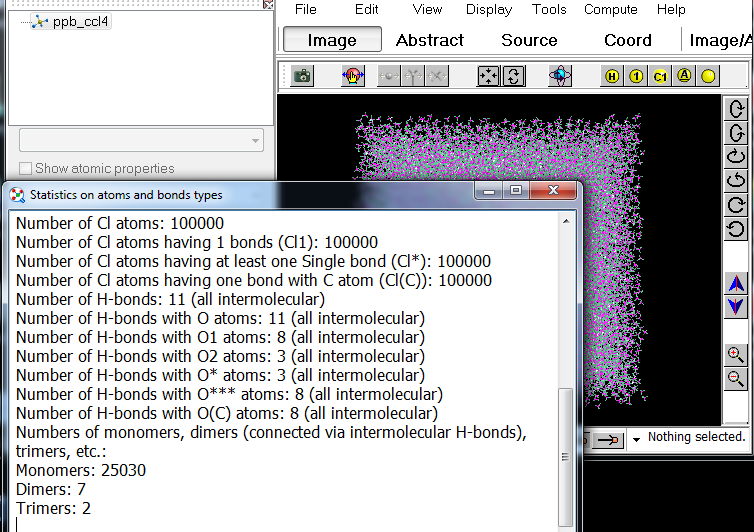
3. Chemcraft can show statistics on the number of H-bonds, including different types of H-bonds - e.g. for O..H bonds with O atoms having one double bound with other atom, etc.:
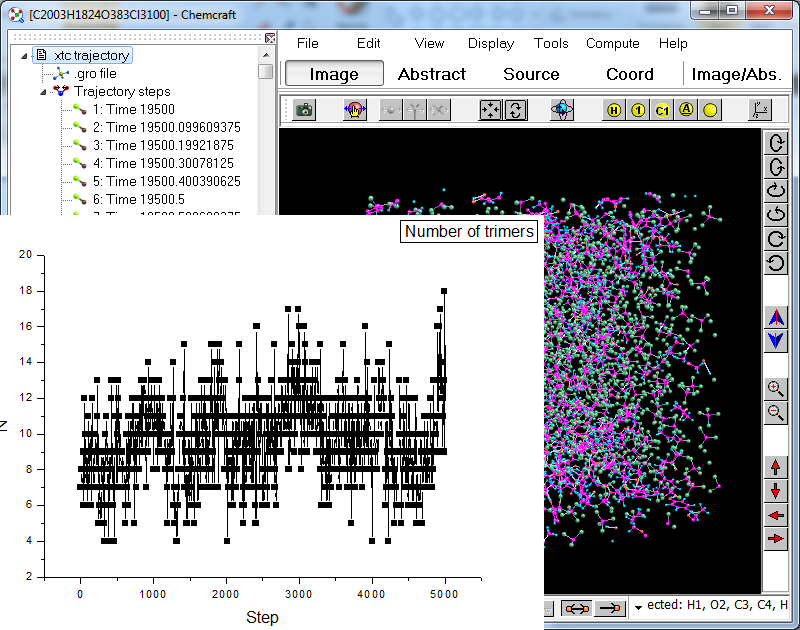
4. The tools above can be used not only for individual trajectory step, but also for all trajectory steps - you can draw a graph of the number of trimers vs trajectory step:
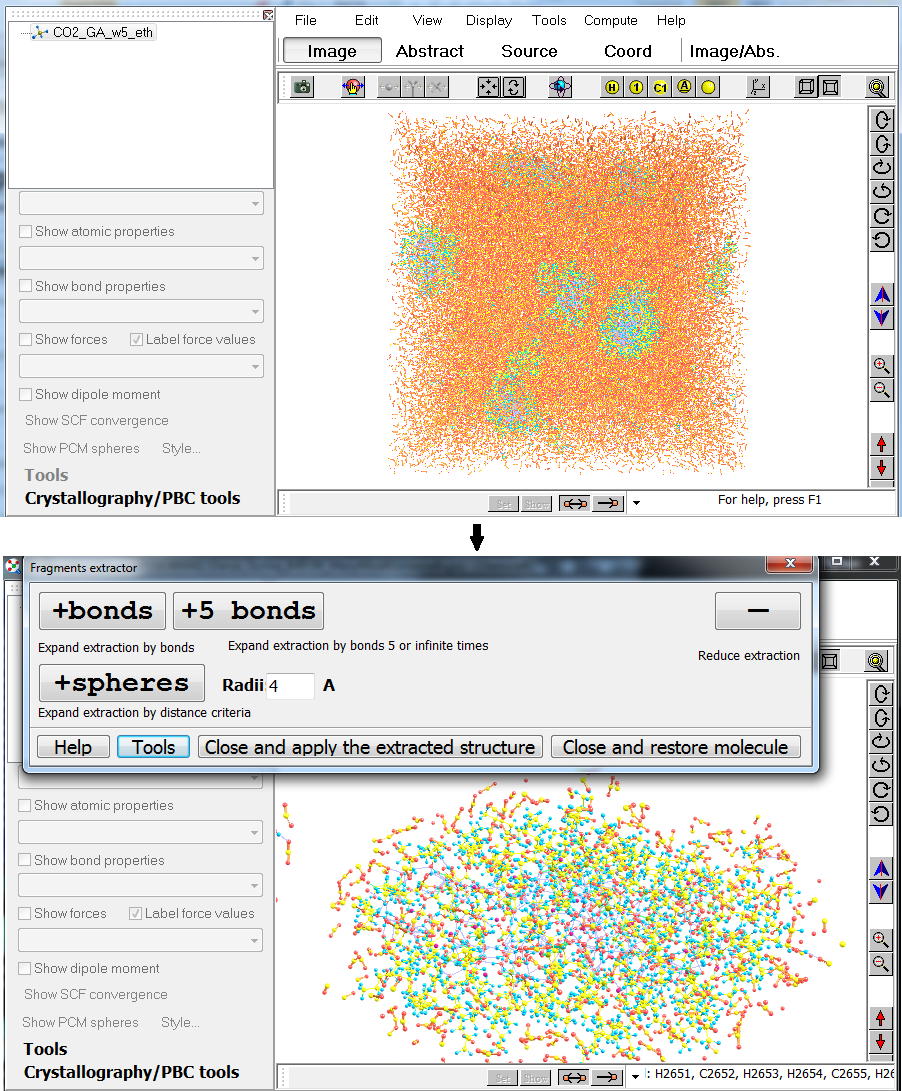
5. Chemcraft has the "Fragments Extractor" tool for both crystallography and molecular dynamics calculations (with PBC cells): firstly the program duplicates the atoms in your cell to build 3*3*3 supercell, then extracts a fragment of this supercell by atoms you have selected. For example, if you have a system containing several micelles in a solvent, you can firstly select all micelles via "Tools/Divide current structure tool", then select one of the micelles via "Edit/Flexible selection options/Selection with rectangle options/Keep selected...", and then apply the Fragments Extractor ("Crystallography/PBC tools/Additional/Show structural subunit...") so you will be able to see only the atoms of the chosen micelle and adjacent atoms:
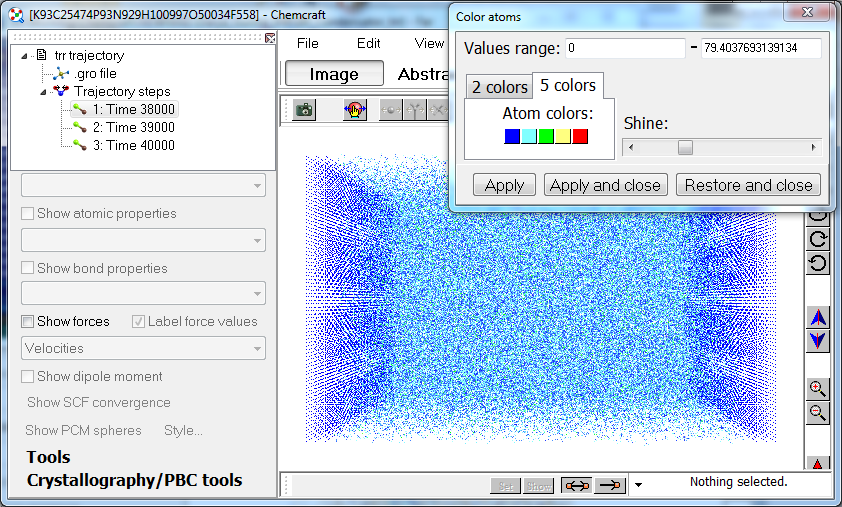
6. Chemcraft can compute the velocities in all steps in your trajectory as the difference between the coordinates of atoms in adjacent steps. The forces can be computed as well;
7. Both for velocities or forces read from Gromacs .trr files or computed by Chemcraft, they can be visualized in the form of atomic colors:
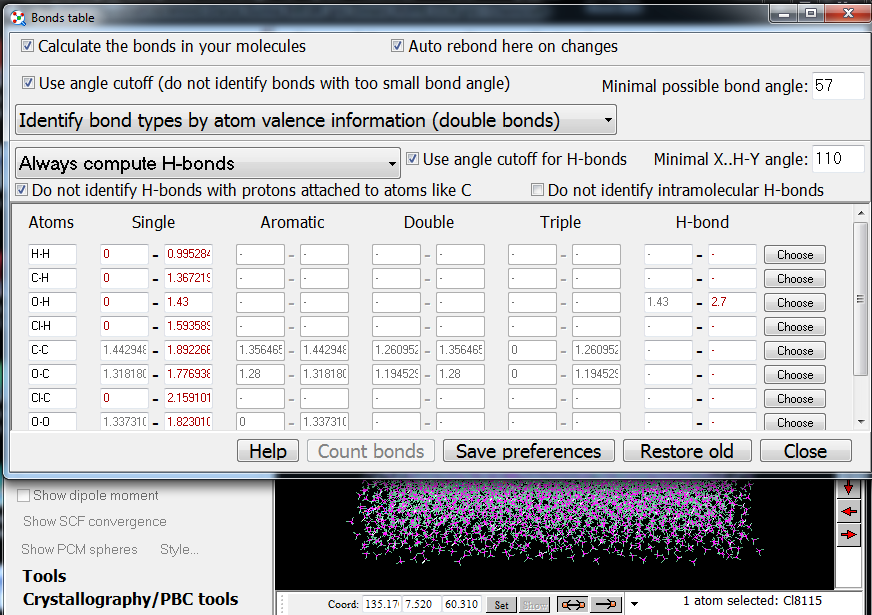
8. Molecular dynamics researchers often need to analyze the network of H-bonds in their systems (e.g. the number of dimers formed by H-bonds). With Chemcraft, such analysis can be made more efficient with manual setting of the parameters used to identify the H-bonds - distance and angle criteria, an option to exclude intermolecular H-bonds or the H-bonds with H atoms attached to atoms like carbons, etc.:
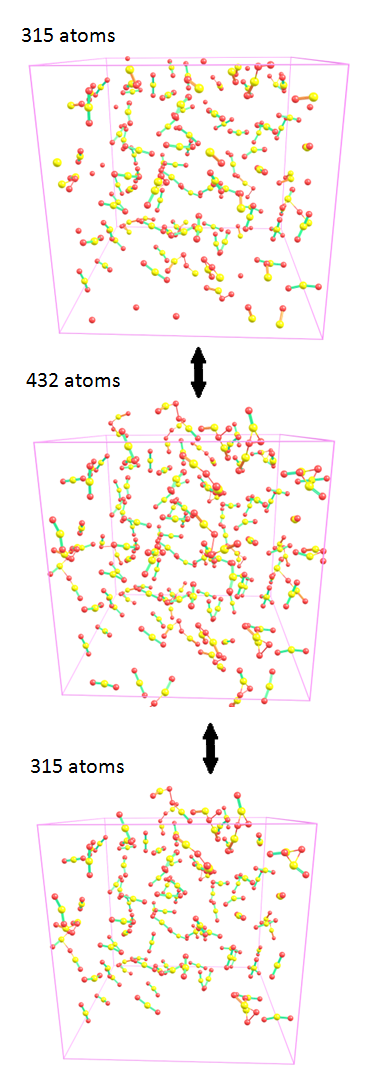
9. Possibly some molecular dynamics researchers can find useful these utilities: either expand a cell to make cropped residues (molecules) at cell borders non-cropped, or vise versa, cut the atoms beyond the cell borders (more exactly, not atoms but whole residues), but keeping only unique residues. In other words, you will be able to switch between three structures: cell with cut residues, e.g. 10 000 atoms, a cell with expanded residues which does not contain any non-whole molecule, e.g. 10 100 atoms, and the expanded cell where some residues containing atoms outside the cell, are removed - again 10 000 atoms:
| |||||||