|
|
|||||
 |
 |
 |
 |
 |
 |
|
ChemCraft basics
ChemCraft comprises a set of graphical tools for facilitating
working with quantum-chemical calculations. It provides convenient
utilities, which help to prepare new jobs for calculation and analyze
computed results. Among the main functions of the program is
visualization of output files produced by quantum-chemical packages. For
this time, the main supported packages are GAMESS (US version) and Gaussian94-03.
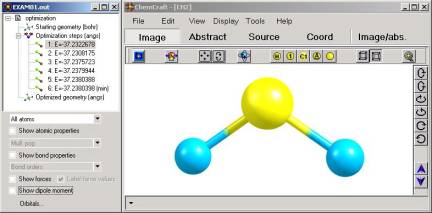
1. Interface of ChemCraft. ChemCraft for Gamess usersChemCraft
provides very detailed visualization of Gamess-US output files. The
following data from the files can be presented graphically: -
Atomic
coordinates (corresponding to either all or symmetry unique atoms, if
corresponding tables are presented in the file); -
If bond
order analysis is presented in the file, bonds from the file are shown
on the image (otherwise, bonds are calculated by distance algorithm); -
Energy
gradient can be shown in the form of pointers (fig. 2); -
Different
atomic properties can be shown as labels on atoms: mulliken populations
and charges, spin densities, valences; -
Bond
orders (as labels on bonds); -
Molecular
orbitals are visualized as isosurfaces or colored planes; -
Vibrational
modes can be animated or shown in the form of pointers (displacement
vectors); -
Dipole
moment can be visualized as pointer. The
program provides structured presentation of output files. The file being
read is divided into separate elements, such as individual geometries or
vibrational modes. For each element all available data is extracted from
the file: atomic coordinates, energy gradient, etc. All elements are
presented in hierarchical list (see fig. 1). Clicking on the elements of
the list automatically displays individual geometries or modes on the
image and allows to visualize different properties. This interface
provides reliable visualization of computational data, including
non-standard types of calculation, incomplete calculations, etc. It also
allows to visualize complicated files with multiple calculation jobs.
For IRC-calculations, all geometries are divided into groups by IRC-steps.
Besides graphical presentation of data, ChemCraft outlines most
essential parts of output file and shows a brief �abstract� for each
element of the file.
2. Example of energy gradient visualization. ChemCraft
extracts molecular orbital coefficients together with basis set
information from Gamess output files and renders molecular orbitals in
the form of isodensity surfaces or
surfaces (planes, spheres) colored by density value (see fig. 3).
ChemCraft provides some possibilities to perform operations with
orbitals (e.g. to multiply one orbital by another). The formulas for
building the orbitals (creation) are taken from the source code of
program PLTORB. The calculation of density values is well-optimized.
Note that if there are several tables of molecular orbital coefficients
in the file, ChemCraft extracts each of them and allows them to be
rendered (for instance, in MCSCF calculations either canonical or
natural orbitals can be shown). Besides visualization of orbitals,
ChemCraft provides a simple utility for automatic determination of
atomic orbitals forming each molecular orbitals, which can be useful for
analysis of the orbitals.
3. Examples of molecular orbitals visualization. ChemCraft
supports an interface to quickly create sections of Gamess input files
with non-standard basis sets (fig. 4). The basis sets are extracted from
their description, which can be obtained at PNNL's webpage (http://www.emsl.pnl.gov/forms/basisform.html). They can be
also supplemented with additional gaussians specified by the user.
4. Basis set creation form. ChemCraft for Gaussian usersWe
recommend to type #P GFINPUT POP(FULL, NBO) in Gaussian input files for
visualization of Gaussian output files via ChemCraft. #P option enables
extended printout; GFINPUT option enables printout of basis set
information (description of primitives in basis set), while POP(FULL)
enables printout of all molecular orbitals coefficients (POP(REGULAR)
can be also used). The last two keywords allow ChemCraft to visualize
molecular orbitals. POP(NBO) enables printout of Natural Bond Orbitals
analysis, in which the bonds in molecule are computed. All these
keywords are advisable but not necessary. As for Gamess files, different
data from the file can be visualized: forces on nucleus (energy
gradient), atomic charges, spin densities and other atomic properties,
NBO bond properties (occupations, energies), normal modes, molecular orbitals
(either Cartesian (6d, etc) or internal (5d) functions can be
visualized). Coordinates in either standard or input/Z-matrix
orientation can be read from the file and shown on the image (it is
necessary for correct visualization of forces on nucleus, because they
are usually printed in different orientation than other properties). For
energy surface scan and IRC jobs, all geometries are grouped by scan
steps. For each individual geometry or vibrational mode, most essential
data is outlined and shown as an �abstract� (SCF energy, convergence
criteria, etc). ChemCraft reads multi-step jobs and presents then as the
list of several expanding nodes, each node representing individual job
in the file. In addition to Gaussian output files, ChemCraft can read Formatted Checkpoint files (.fch), extracting molecular structure and orbitals from the file. For visualization of molecular orbitals and other properties, Gaussian Cube files can be also read. ChemCraft
reads isotropic shielding values from Gaussian log files with NMR
calculations (GIAO, CSGT). A simple utility for recounting them into
chemical shifts and averaging within specified groups of atoms is
provided. Working with other formatsBesides
Gamess and Gaussian files, ChemCraft can read HyperChem files, files of
MSI or PDB formats (these formats are not comprehensively supported),
MolDraw and Priroda programs format, ADF ASCII TAPE41 files, and simple text files with
Cartesian coordinates of atoms. ChemCraft provides an interface to
import/export coordinates of atoms in text format through clipboard,
which helps to use data from any types of calculations. Export of atomic
coordinates into clipboard can be also useful for creating input files. ChemCraft comprises an utility for conversion of fractional coordinates, used in crystallographic measurements, into Cartesian, and conversely, using unit cell parameters (a,b, etc). Constructing molecules
ChemCraft supports a set of tools for constructing molecular
structures, which can be used for preparing an initial guess for
calculations and other purposes: -
Constructing
molecule from standard molecular fragments (radicals, etc). The
possibility to supplement the set of fragments with custom ones and to
copy/paste individual fragments via clipboard is provided; -
Modifying
any geometrical parameter in the molecule (distance, angle, dihedral).
The modification can be accompanied with displacement of one atom, two
atoms or selected group of atoms; -
The
possibility to �drag� an atom or a fragment on the molecule�s
image or rotate a fragment using the mouse (fig. 5); -
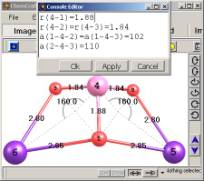
Iterative
algorithm for applying an arbitrary set of geometrical parameters (fig.
6); - An easy to use utility for applying a point group to the molecule.
5. Examples of �dragging� an atom or rotating a fragment along a bond. When
�dragging� an atom or performing other structural modifications, any
geometrical parameter can be controlled on the image (see fig. 5).
ChemCraft�s interface allows to easily alter the type of any atom or
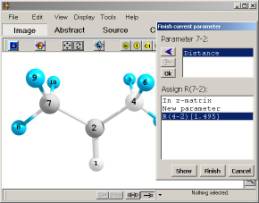
insert/remove a bond. ChemCraft
provides an utility for quick obtaining z-matrix. The z-matrix is build
by clicking on atoms in molecule and specifying some additional
information (fig. 7). Before using this utility one should first obtain
the structure of molecule in Cartesian coordinates. All above-mentioned
tools for constructing molecules can be used for this purpose.
6. Applying a specified set of geometrical parameters.
7. Visual construction of Z-matrix. Molecule rendering possibilities
ChemCraft produces high-quality 32-bit pictures of molecules. It
is designed as a program for creating publication-ready images, which
does not require any additional modification. The pictures can be easily
supplemented with captions on atoms/bonds and additional objects, such
as labels and lines. ChemCraft comprises a collection of standard
graphical schemes. Each scheme represents a set of parameters defining
the appearance of the molecule: lighting parameters, colors and sizes of
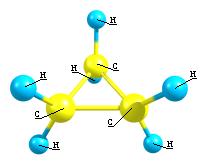
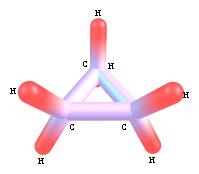
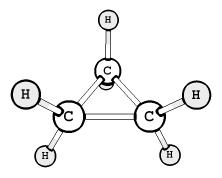
individual atoms and bonds, etc. Fig. 8 illustrates four schemes of the
collection. ChemCraft allows the user to change the parameters of
individual schemes or add his own schemes to the collection. The
graphical engine of ChemCraft does not require any graphical
acceleration either as additional graphical libraries. It is
well-optimized and provide high rendering speed even on outdated
computers.
8. Examples of rendering a molecule in different graphical schemes and with different labels style.
|
||||||